Cornelia de Lange syndrome: A rare genetic disorder
Baburao Sonawane V.1*, Kotrashetti V.2, Bainade K.3, Vatkar A.4, Pansare A.5
DOI: https://doi.org/10.17511/ijpr.2020.i03.07
1* Vijay Baburao Sonawane, Associate Professor, Department of Pediatrics, D.Y. Patil University, School of Medicine, Nerul, Maharashtra, India.
2 V. Kotrashetti, Professor and Head of Unit, Department of Pediatrics, D.Y. Patil University, School of Medicine, Nerul, Maharashtra, India.
3 Kapil Bainade, Assistant Professor, Department of Pediatrics, D.Y. Patil University, School of Medicine, Nerul, Maharashtra, India.
4 Amit Vatkar, Assistant Professor, Department of Pediatrics, D.Y. Patil University, School of Medicine, Nerul, Maharashtra, India.
5 Abolee Pansare, Resident (PG), Department of Pediatrics, D.Y. Patil University, School of Medicine, Nerul, Maharashtra, India.
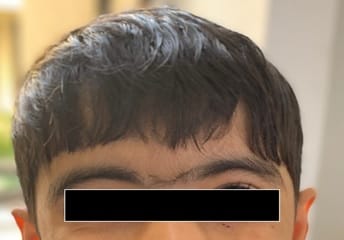
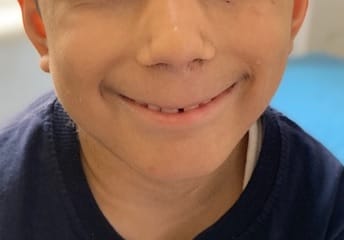
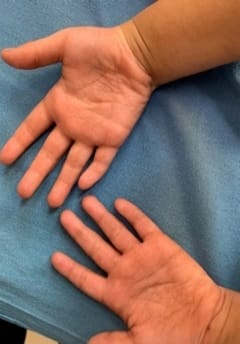
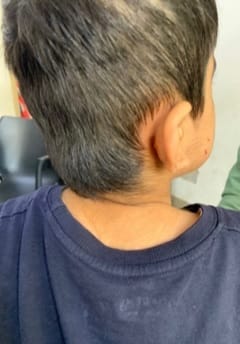
Cornelia de Lange Syndrome (CdLS) was first reported by Vrolik in 1849 and Brachmann in 1916, followed by Cornelia de Lange in 1933, after whom the syndrome is named. This disorder has a varied presentation but is mainly characterized by distinctive facial features, growth retardation, microcephaly, hirsutism, psychomotor delay, intellectual disability, and malformations of the upper limbs. Initial diagnosis is usually based on clinical features following specific diagnostic scoring systems. The precise prevalence of the disease is unknown but is estimated to be 1–10:100,000. Depending on the mutated gene, Cornelia de Lange syndrome (CdLS) can be inherited in an autosomal dominant manner, when it is caused by variations in the NIPBL, SMC2, or RAD21 genes, or it can have an X-linked inheritance when it is caused by variations in the SMC1A or HDAC8 genes. However, most cases (more than 99%) result from new (de novo) mutations, which means that are not inherited from the parents and occur in people with no family history of the conditionabout 30% of the people affected by the syndrome do not have any known cause. Many studies focused on the importance of neurologic findings and reported an incidence of epilepsy in CdLS ranging from 14% to 25%, especially in the classic and more severe form of the syndrome, but there is no data about its electroclinical features and long-term outcome. Life expectancy is relatively normal for people with CdLS and most affected children live well into adulthood. However, certain features of this condition, particularly severe malformations of the heart or throat, may decrease life expectancy in some affected people. The diagnosis is suspected clinically and later confirmed by clinical exome sequencing.
Keywords: Cornelia de Lange Syndrome (CdLS), Epilepsy, Developmental Delay, Clinical Exome Sequencing
| Corresponding Author | How to Cite this Article | To Browse |
|---|---|---|
| , Associate Professor, Department of Pediatrics, D.Y. Patil University, School of Medicine, Nerul, Maharashtra, India. Email:  |
Sonawane VB, Kotrashetti V, Bainade K, Vatkar A, Pansare A. Cornelia de Lange syndrome: A rare genetic disorder. Pediatric Rev Int J Pediatr Res. 2020;7(3):152-156. Available From https://pediatrics.medresearch.in/index.php/ijpr/article/view/569 |
 |
 ©
©