William Syndrome Presenting with Life-threatening Heart failure: Rare case report
Mehrotra A.1*, Shakya U.2, Kacker S.3
DOI: https://doi.org/10.17511/ijpr.2023.i03.05
1* Akhil Mehrotra, Chief, Non Invasive Cardiologist Pediatric and Adult Cardiology, Prakash Heart Station, D-16 Nirala Nagar, Lucknow, Uttar Pradesh, India.
2 Ujala Shakya, Cardiac Technician, Prakash Heart Station, D-16 Nirala Nagar, Lucknow, Uttar Pradesh, India.
3 Shubham Kacker, Lead PMO, Tech Mahindra, New Delhi, India.
Williams syndrome (WS), also referred to as Williams-Beuren syndrome, is a rare complex congenital developmental multisystem disorder, occurring in 1 per 20,000 live births, It is characterized by congenital heart defects (CHD), skeletal, renal anomalies, cognitive disorder, social personality disorder and notably dysmorphic Elfin-like facies. Supravalvular aortic stenosis is the most frequent cardiovascular abnormality in WS children. WS occurs as the result of a deletion of approximately 1.5-1.8 Mb on chromosome 7q11.23. The deletion is almost always denovo, however, familial cases have been reported. A genetic study is usually required for a definitive diagnosis, but genetic testing is often unavailable in developing countries and the combination of a typical clinical phenotype and echocardiographic profile helps to confirm the diagnosis. We are reporting a rare case of WS in a 2-month infant presenting with heart failure because of multiple CHDs, including coarctation of the aorta (COA), patient ductus arteriosus (PDA), and mild supravalvular pulmonary stenosis (SVPS), severe-pulmonary hypertension and systemic hypertension.
Keywords: Heart failure, COA, PDA, Supra-Valvular Pulmonary Stenosis, Elfin Facies
| Corresponding Author | How to Cite this Article | To Browse |
|---|---|---|
| , Chief, Non Invasive Cardiologist Pediatric and Adult Cardiology, Prakash Heart Station, D-16 Nirala Nagar, Lucknow, Uttar Pradesh, India. Email:  |
Akhil Mehrotra, Ujala Shakya, Shubham Kacker, William Syndrome Presenting with Life-threatening Heart failure: Rare case report. Pediatric Rev Int J Pediatr Res. 2023;10(3):57-64. Available From https://pediatrics.medresearch.in/index.php/ijpr/article/view/751 |
 |
 ©
© 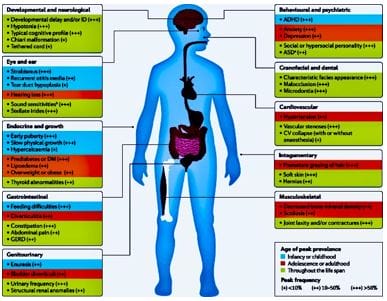 Figure 1: Williams’ Syndrome - myriads of clinical features
Figure 1: Williams’ Syndrome - myriads of clinical features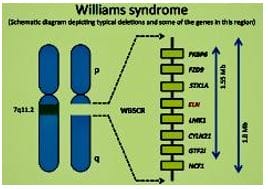 Figure 2: There is microdeletion on chromosome 7, resulting in the loss of 26-28 genes.
Figure 2: There is microdeletion on chromosome 7, resulting in the loss of 26-28 genes.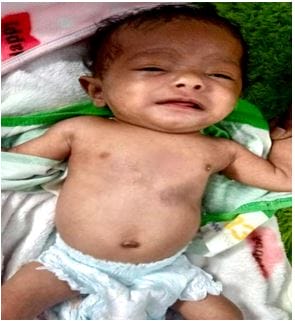 Figure 3: severely dyspneic, irritable child, with obvious chest retractions and pectus excavatum deformity.
Figure 3: severely dyspneic, irritable child, with obvious chest retractions and pectus excavatum deformity. Figure 4: Typical Elfin facies: wide forehand, bilateral epicanthal folds, wide-set eyes, sunken bridge of the nose, wide nostrils with upturned nose, full cheeks, pointed chin, large right ear, left ear having cup deformity.
Figure 4: Typical Elfin facies: wide forehand, bilateral epicanthal folds, wide-set eyes, sunken bridge of the nose, wide nostrils with upturned nose, full cheeks, pointed chin, large right ear, left ear having cup deformity.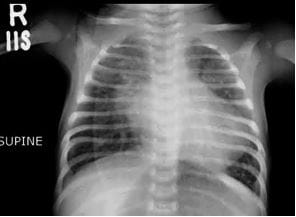 Figure 5: Chest Xray PA view Cardiomegaly, Biatrial enlargement, Pulmonary Venous congestion, Pulmonary
Figure 5: Chest Xray PA view Cardiomegaly, Biatrial enlargement, Pulmonary Venous congestion, Pulmonary
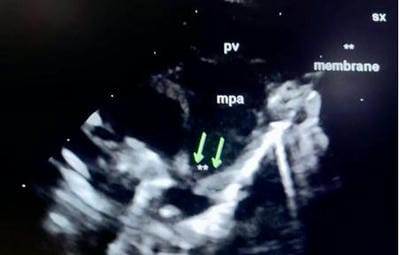 Figure 7: supravalvular pulmonary stenosis- In the SX view, a constriction band is visualised just above the bifurcation of the main pulmonary artery. The pulmonary valve annulus and main pulmonary artery are dilated and the left and right branch pulmonary arteries are of normal size: pv annulus (D) 14.2 mm, main pulmonary artery (D) 13.4 mm, left pulmonary artery (D) 4.9 mm, right pulmonary artery (D) 4.6 mm
Figure 7: supravalvular pulmonary stenosis- In the SX view, a constriction band is visualised just above the bifurcation of the main pulmonary artery. The pulmonary valve annulus and main pulmonary artery are dilated and the left and right branch pulmonary arteries are of normal size: pv annulus (D) 14.2 mm, main pulmonary artery (D) 13.4 mm, left pulmonary artery (D) 4.9 mm, right pulmonary artery (D) 4.6 mm Figure 8: supravalvular pulmonary stenosis: On CW analysis peak/mean gradient across the supravalvular stenotic band was 17.7/9 mmhg, corresponding with mild supravalvular stenosis
Figure 8: supravalvular pulmonary stenosis: On CW analysis peak/mean gradient across the supravalvular stenotic band was 17.7/9 mmhg, corresponding with mild supravalvular stenosis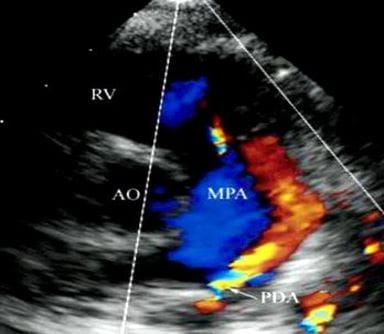 Figure 9: PDA - In the SX view on color flow doppler analysis a continuous flow (red) from the PDA into the main pulmonary artery is displayed. AO, aorta, RV, right ventricle, MPA, main pulmonary artery
Figure 9: PDA - In the SX view on color flow doppler analysis a continuous flow (red) from the PDA into the main pulmonary artery is displayed. AO, aorta, RV, right ventricle, MPA, main pulmonary artery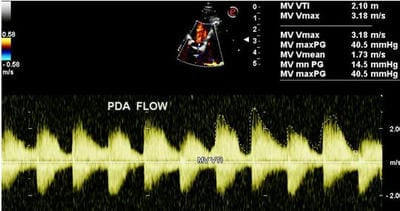 Figure 10: On CW Doppler analysis across PDA peak/mean gradient was 40.5/ 14.5 MMHg
Figure 10: On CW Doppler analysis across PDA peak/mean gradient was 40.5/ 14.5 MMHg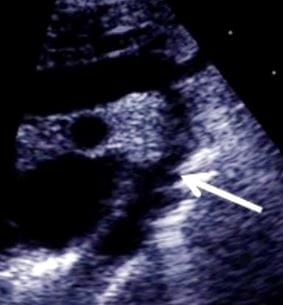 Figure 11: suprasternal view: The arrow depicts the site of COA, just beyond the origin of the left subclavian artery.
Figure 11: suprasternal view: The arrow depicts the site of COA, just beyond the origin of the left subclavian artery.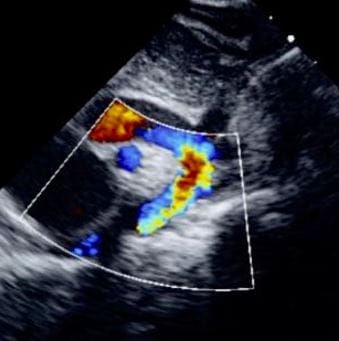 Figure 12: Suprasternal View demonstrates a turbulent mosaic pattern flow in the region of COA and descending aorta.
Figure 12: Suprasternal View demonstrates a turbulent mosaic pattern flow in the region of COA and descending aorta. Figure 13: CW Doppler flow across COA is indicative of a peak/ mean gradient of 43.5/25 mm hg, consistent with moderate COA
Figure 13: CW Doppler flow across COA is indicative of a peak/ mean gradient of 43.5/25 mm hg, consistent with moderate COA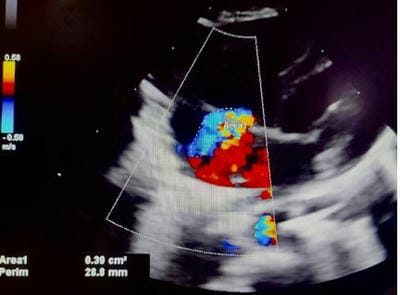 Figure 14: 4CH View: A turbulent, moderate tricuspid regurgitation jet (blue) is recognized in the right atrium.
Figure 14: 4CH View: A turbulent, moderate tricuspid regurgitation jet (blue) is recognized in the right atrium.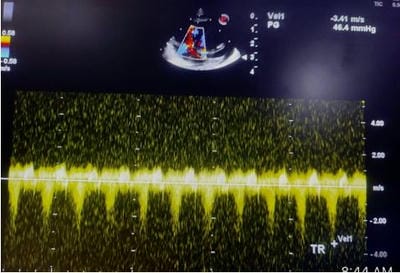 Figure 15: CW Doppler across Tricuspid Regurgitation jet shows a peak velocity of 3.6 m/ sec (gradient 52 mm hg). The estimated right ventricular systolic pressure/ pulmonary artery pressure was (52+ 15) 67 mm hg, suggestive of severe pulmonary hypertension.
Figure 15: CW Doppler across Tricuspid Regurgitation jet shows a peak velocity of 3.6 m/ sec (gradient 52 mm hg). The estimated right ventricular systolic pressure/ pulmonary artery pressure was (52+ 15) 67 mm hg, suggestive of severe pulmonary hypertension.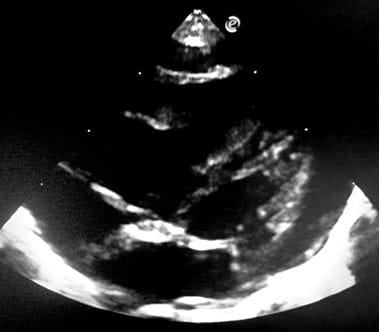 Figure 16: modified 4CH view: delineates dilatation of right ventricular and right atrium.
Figure 16: modified 4CH view: delineates dilatation of right ventricular and right atrium.