An Atypical Presentation of Pyridoxine Dependent Epilepsy
Susarla B1*, Chintapally S2, Gundapuneni R3, Gattu H4
DOI: https://doi.org/10.17511/ijpr.2023.i06.02
1* Balaji Venkata Rama Susarla, Consultant, Department Of Neonatology, Ankura Hospital For Women And Children, Boduppal, Hyderabad, Telangana, India.
2 Suman Kumar Chintapally, Consultant, Department Of Neonatology, Ankura Hospital for Women and Children, Boduppal, Hyderabad, Telangana, India.
3 Ravali Gundapuneni, Consultant, Department Of Neonatology, Ankura Hospital for Women and Children, Boduppal, Hyderabad, Telangana, India.
4 Harshita Gattu, Consultant, Department Of Neonatology, Ankura Hospital for Women and Children, Boduppal, Hyderabad, Telangana, India.
Pyridoxine-dependent epilepsy (PDE) is a rare autosomal recessive disorder and is considered a prototypical form of metabolic epilepsy. It is characterised by recurrent seizures in the prenatal, neonatal and/or postnatal periods that are resistant to conventional antiepileptic drugs, but responsive to pharmacological doses of pyridoxine. The point prevalence of PDE may vary from 1:20,000 to 1:600,000, based on the degree of systemic ascertainment via therapeutic trial with pyridoxine. Often, pyridoxine-dependent epilepsy can cause diagnostic dilemmas, due to a lack of awareness of this clinical condition, the wide spectrum of clinical manifestations and phenotype genotype variances. Accumulation of the toxic metabolites will lead to developmental delay and mental retardation in the long term, which can be prevented by pyridoxine supplementation and restriction of lysine from diet. Hence, early detection of this condition is very important. Novel instrumental screening methods need to be developed for early detection of this condition to achieve better seizure control and prevent long-term brain damage in this group of patients. Here, we report a case of a newborn who presented with atypical features of neonatal epileptic encephalopathy along with abdominal distension and a mixed seizure pattern and was diagnosed as pyridoxine-dependent epilepsy on whole exome sequencing.
Keywords: Pyrodoxine, deficiency, Epilepsy, Epilepsy
| Corresponding Author | How to Cite this Article | To Browse |
|---|---|---|
| , Consultant, Department Of Neonatology, Ankura Hospital For Women And Children, Boduppal, Hyderabad, Telangana, India. Email:  |
Susarla B, Chintapally S, Gundapuneni R, Gattu H, An Atypical Presentation of Pyridoxine Dependent Epilepsy. Pediatric Rev Int J Pediatr Res. 2023;10(6):101-104. Available From https://pediatrics.medresearch.in/index.php/ijpr/article/view/760 |
 |
 ©
© 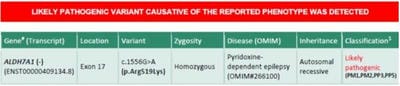
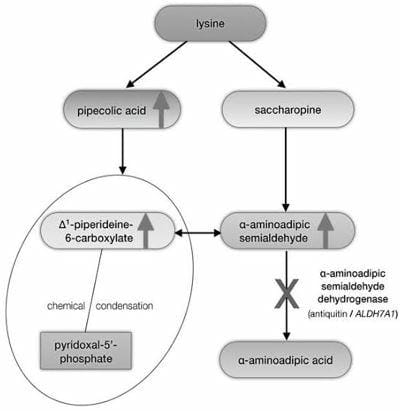 Figure 2: Pathophysiology of pyridoxine dependent epilepsy showing the accumulation of toxic metabolites.Pyridoxine supplementation compensates for the chemical pyridoxal 5 phosphate inactivation, but the accumulation of substrates from lysine degradation is not sufficiently reduced.
Figure 2: Pathophysiology of pyridoxine dependent epilepsy showing the accumulation of toxic metabolites.Pyridoxine supplementation compensates for the chemical pyridoxal 5 phosphate inactivation, but the accumulation of substrates from lysine degradation is not sufficiently reduced.