Krabbe disease - a rare lysosomal storage disease
Baburao Sonawane V.1, Kotrashetti V.2, S Bainade K.3, Vatkar A.4, R Bhatarkar S.5*
DOI: https://doi.org/10.17511/ijpr.2020.i05.05
1 Vijay Baburao Sonawane, Associate Professor, Department of Pediatrics, D.Y. Patil University, School of Medicine, Nerul, Maharashtra, India.
2 V.A. Kotrashetti, Professor and Head of Unit, Department of Pediatrics, D.Y. Patil University, School of Medicine, Nerul, Maharashtra, India.
3 Kapil S Bainade, Assistant Professor, Department of Pediatrics, D.Y. Patil University, School of Medicine, Nerul, Maharashtra, India.
4 Amit Vatkar, Assistant Professor, Department of Pediatrics, D.Y. Patil University, School of Medicine, Nerul, Maharashtra, India.
5* Shuchi R Bhatarkar, Junior Resident (PG), Department of Pediatrics, D.Y. Patil University, School of Medicine, Nerul, Maharashtra, India.
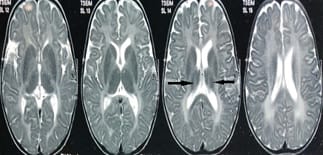
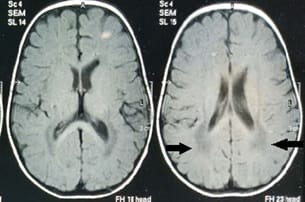
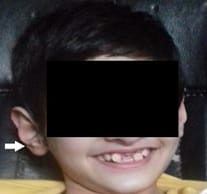
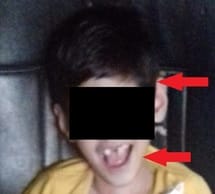
Krabbe disease a rare and fatal lysosomal storage disease that results in progressive damage, demyelination of central, and peripheral nervous system. It involves the dysfunctional metabolism of sphingolipids; characterized by the deficiency of enzyme galactocerebrosidase (galactosylceramidase, GALC). Krabbe disease is an extremely rare condition with an incidence of 1 in 1,00,000 live births. Typically, the disease has an infantile-onset, with rapid deterioration in the first few months, leading to death before the age of 2 years. The late-onset forms (late-infantile, juvenile, and adult forms) are rare with variable clinical outcomes, presenting spastic paraplegia as the main symptom. It is inherited in an autosomal recessive pattern. Early diagnosis of Krabbe disease can be made by measuring the activity of galactocerebrosidase in a sample of a dried spot of blood during the screening of new-born infants. Radiological investigations can show diffuse brain and cerebellar atrophy, and demyelination can be identified by magnetic resonance imaging (MRI). Authors report a case of a 10-year-old female child presented with feeding difficulties excessive irritability, global developmental delay, and progressive loss of hearing and sight. On examination had peculiar facial features, hypertonia, and hyper reflexes. No neurocutaneous stigmata were found. Diagnosed as Krabbe's disease on the basis of peculiar clinical features and confirmed diagnosis on MRI and spectrophotometric and spectrofluorometric enzyme assay. There's no cure for Krabbe disease, and treatment focuses on supportive care. However, stem cell transplants have shown some success in infants who are treated before the onset of symptoms and in some older children and adults.
Keywords: Krabbe disease, Autosomal recessive, Progressive neurologic degeneration, GALC, Globoid cell leukodystrophy, Enzyme assay
| Corresponding Author | How to Cite this Article | To Browse |
|---|---|---|
| , Junior Resident (PG), Department of Pediatrics, D.Y. Patil University, School of Medicine, Nerul, Maharashtra, India. Email:  |
Sonawane VB, Kotrashetti VA, Bainade KS, Vatkar A, Bhatarkar SR. Krabbe disease - a rare lysosomal storage disease. Pediatric Rev Int J Pediatr Res. 2020;7(5):217-221. Available From https://pediatrics.medresearch.in/index.php/ijpr/article/view/602 |
 |
 ©
©