
Krabbe disease - a rare lysosomal storage disease
Abstract
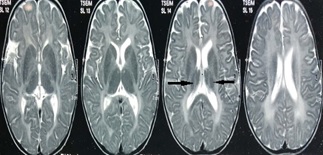
Krabbe disease a rare and fatal lysosomal storage disease that results in progressive damage, demyelination of central, and peripheral nervous system. It involves the dysfunctional metabolism of sphingolipids; characterized by the deficiency of enzyme galactocerebrosidase (galactosylceramidase, GALC). Krabbe disease is an extremely rare condition with an incidence of 1 in 1,00,000 live births. Typically, the disease has an infantile-onset, with rapid deterioration in the first few months, leading to death before the age of 2 years. The late-onset forms (late-infantile, juvenile, and adult forms) are rare with variable clinical outcomes, presenting spastic paraplegia as the main symptom. It is inherited in an autosomal recessive pattern. Early diagnosis of Krabbe disease can be made by measuring the activity of galactocerebrosidase in a sample of a dried spot of blood during the screening of new-born infants. Radiological investigations can show diffuse brain and cerebellar atrophy, and demyelination can be identified by magnetic resonance imaging (MRI). Authors report a case of a 10-year-old female child presented with feeding difficulties excessive irritability, global developmental delay, and progressive loss of hearing and sight. On examination had peculiar facial features, hypertonia, and hyper reflexes. No neurocutaneous stigmata were found. Diagnosed as Krabbe's disease on the basis of peculiar clinical features and confirmed diagnosis on MRI and spectrophotometric and spectrofluorometric enzyme assay. There's no cure for Krabbe disease, and treatment focuses on supportive care. However, stem cell transplants have shown some success in infants who are treated before the onset of symptoms and in some older children and adults.
Downloads
References
Vargiami E, Papathanasiou E, Batzios S, Kyriazi M, Dimitriou E, Anastasiou A, et al. Balkan. Neuroradiological, neurophysiological and molecular findings in infantile Krabbe disease: two case reports. Med Genet. 2016;19(1):85-90. doi: https://dx.doi.org/10.1515%2Fbjmg-2016-0011.
White AB, Givogri MI, Lopez-Rosas A, Cao H, van Breemen R, Thinakaran G, et al. Psychosine accumulates in membrane microdomains in the brain of Krabbe patients, disrupting the raft architecture. Neurosci. 2009;29(19):6068-6077. doi: https://doi.org/10.1523/jneurosci.5597-08.2009.
Sakai N, Inui K, Fujii N, Fukushima H, Nishimoto J, Yanagihara I, et al. Krabbe disease: isolation and characterisation of a full-length cDNA for human galactocerebrosidase. Biochem Biophys Res Commun. 1994;198(2):485–491. doi: https://doi.org/10.1006/bbrc.1994.1071.
Thomas PK, Halpern JP, King RH, Patrick D. Galactosylceramide lipidosis: novel presentation as a slowly progressive spinocerebellar degeneration. Ann Neurol. 1984;16(5):618-620. doi: https://doi.org/10.1002/ana.410160515.
Loonen MC, Van Diggelen OP, Janse HC, Kleijer WJ, Arts WF. Late-onset globoid cell leukodystrophy (Krabbe’s disease). Clinical and genetic delineation of two forms and their relation to the early-infantile form. Neuropediatrics. 1985;16(3):137-142. doi: https://doi.org/10.1055/s-2008-1052558.
Grover S B, Gupta P, Jain M, Kumar A, Gulati P. Characteristic CT and MR features of Krabbe's disease: A case report. Indian J Radiol Imaging. 2005;15(4):503-506. Available from: http://www.ijri.org/text.asp?2005/15/4/503/28783.
Debs R, Froissart R, Aubourg P, Papeix C, Douillard C, Degos B, et al. Krabbe disease in adults: Phenotypic and genotypic update from a series of 11 cases and a review. Inherit Metab Dis. 2013;36(5):859-868. doi: https://doi.org/10.1007/s10545-012-9560-4.
Hossain MA, Otomo T, Saito S, Ohno K, Sakuraba H, Hamada Y, et al. Late-onset Krabbe disease is predominant in Japan and its mutant precursor protein undergoes more effective processing than the infantile-onset form. Gene. 2014;534(2):144-154. doi: https://doi.org/10.1016/j.gene.2013.11.003.
Krabbe K. A new familial, infantile form of diffuse brain-sclerosis. Brain. 1916;39(1-2):74-114.
Suzuki K, Suzuki Y. Krabbe’s globoid cell leukodystrophy: Deficiency of galactocerebroside beta-galactosidase activity. Neuropathol Exp Neurol. 1971;30(1):145.
Wenger DA, Sattler M, Clark C, McKelvey H. An improved method for the identification of patients and carriers of Krabbe’s disease. Clin Chim Acta. 1974;56(2):199-206. doi: https://doi.org/10.1016/0009-8981(74)90228-9.
Allewelt H, Taskindoust M, Troy J, Page K, Wood S, Parikh S, et al. Long-term functional outcomes after hematopoietic stem cell transplant for early infantile Krabbe disease. Biol Blood Marrow Transplant. 2018;24(11):2233-2238. doi: https://doi.org/10.1016/j.bbmt.2018.06.020.
Given CA, 2nd, Santos CC, Durden DD. Intracranial and spinal MR imaging findings associated with Krabbe’s disease: Case report. Am J Neuroradiol. 2001;22(9):1782-1785.
Wenger DA. Krabbe disease. In: Pagon RA, Adam MP, Ardinger HH, editors. Gene Reviews. Seattle, WA: p. 1993.
Jalal K, Carter R, Yan L, Barczykowski A, Duffner PK. Does galactocerebrosidase activity predict Krabbe phenotype? Pediatr Neurol. 2012;47(5):324-329. doi: https://doi.org/10.1016/j.pediatrneurol.2012.07.003.
Copyright (c) 2020 Author (s). Published by Siddharth Health Research and Social Welfare Society

This work is licensed under a Creative Commons Attribution 4.0 International License.

 OAI - Open Archives Initiative
OAI - Open Archives Initiative