A novel RET gene mutation in a neonate with total colonic aganglionosis and renal agenesis: a case report
Teja Goud M R.1, Kumar K.2, Banu S.3*
DOI: https://doi.org/10.17511/ijpr.2020.i08.08
1 Ravi Teja Goud M, DNB (Pediatric), Department of Neonatology, Ankura Hospital for Women and Children, Hyderabad, Telangana, India.
2 K. Lalatendu Kumar, Consultant, Pediatric Surgeon and Pediatric Urologist, Ankura Hospital for Women and Children, Hyderabad, Telangana, India.
3* S. Nasreen Banu, Consultant Neonatologist, Department of Neonatology, Ankura Hospital for Women and Children, Hyderabad, Telangana, India.
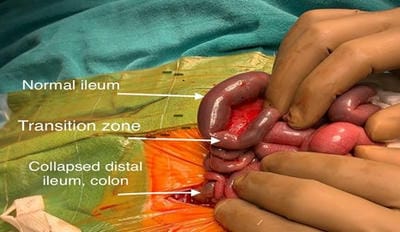
Hirschsprung’s disease (HSCR) is functional lower intestinal obstruction, due to the congenital absence of the intramural plexuses of ganglion cells in the distal bowel. Total colonic aganglionosis (TCA) is a rare and severe form of HSCR and accounts for 5-10% of all the diagnosed cases of HSCR. TCA is a diagnostic and therapeutic challenge as clinical and radiological findings are not pathognomonic. The RET gene signaling system is generally acknowledged as being the most important in TCA pathogenesis, with RET gene variations being present in 70% of cases. The present study is reporting a case of TCA and right renal agenesis with a novel mutation in the RET gene.
Keywords: Hirschsprung’s disease (HSCR), Total colonic aganglionosis (TCA), REarranged during Transfection gene (RET)
| Corresponding Author | How to Cite this Article | To Browse |
|---|---|---|
| , Consultant Neonatologist, Department of Neonatology, Ankura Hospital for Women and Children, Hyderabad, Telangana, India. Email:  |
Teja Goud MR, Kumar KL, Banu SN. A novel RET gene mutation in a neonate with total colonic aganglionosis and renal agenesis: a case report. Pediatric Rev Int J Pediatr Res. 2020;7(8):437-441. Available From https://pediatrics.medresearch.in/index.php/ijpr/article/view/635 |
 |
 ©
©